北京新阳光慈善基金会于2009年在北京市民政局注册成立,其前身为2002年成立的北京大学阳光志愿者协会,由北京大学学生刘正琛在身患白血病后发起成立,2013年在国内率先由非公募基金会转型为公募基金会。新阳光在成立后本着“用爱自己的心去爱别人”的宗旨,在医疗和教育领域创建了“新阳光病房学校”、“生命的礼物”、“阳光骨髓库”等多个品牌项目。

北京新阳光慈善基金会于2009年在北京市民政局注册成立,其前身为2002年成立的北京大学阳光志愿者协会,由北京大学学生刘正琛在身患白血病后发起成立,2013年在国内率先由非公募基金会转型为公募基金会。新阳光在成立后本着“用爱自己的心去爱别人”的宗旨,在医疗和教育领域创建了“新阳光病房学校”、“生命的礼物”、“阳光骨髓库”等多个品牌项目。

关于北京新阳光慈善基金会的新闻信息、通知公告、阳光故事、活动预告和宣传片等信息。欢迎关注我们的信息报道,我们会第一时间提供准确、真实、可信的信息。同时您可以使用网站搜索功能,获得新闻事件、热点话题、人物动态和公益资讯等,快速了解新阳光的最新进展。

为满足患者经济、心理、信息等方面的需求,新阳光为患者提供多个项目,目的是联结基金会内外部资源,为患者及其家庭缓解各方面压力。我们通过向患者拨付救助款缓解患者经济压力,通过开设病房学校让长期住院儿童享受陪伴式教育,通过设立慈善医保补充基金让欠发达地区儿童看得起病,通过资助医生和医学生提高欠发达地区医疗水平,通过开展疾病知识讲座、制作医疗知识手册增强患者和患者家属对医疗知识及护理知识的了解,增进患者群体之间的交流与分享。

新阳光基金会根据捐赠人或发起人意愿设立、实行专款专用、在本组织运作框架下有一定独立自主性的专用资金。目前在新阳光设立的专项基金有: 峥爱基金、 助医儿童白血病研究基金、 一元儿童噬血基金、 儿童舒缓治疗专项基金、 香柏树专项基金、 爱在专项基金、 爱里的心先心病专项基金、 V爱血液病公益基金、 源公益专项基金、 焕蓝梦想基金、 京西专项基金、 信美相互少儿救助专项基金等。

新阳光联劝伙伴(竹林计划)是北京新阳光慈善基金支持的医疗、教育等领域的初创或者草根公益组织,新阳光为这些组织提供团队培训、能力建设、联合劝募、宣传倡导、咨询等服务和支持,致力于促进医疗、教育等领域公益事业的发展,更好地服务目标人群。

关于北京新阳光慈善基金会的工作报告、年检报告、财务报告、内部刊物、财务公示、规章制度、理事会情况等。慈善组织信息公开像绿色蔬菜那样,实现从田间地头到市民餐桌的全面公开链,老百姓拿到一把菜,就可以知道是谁种的,在哪里种的,施肥管理情况如何等等。新阳光的信息公开也是这样,哪怕民众捐一块钱,也知道这一块钱用在了哪里,谁用了,用的效果如何。

欢迎加入新阳光志愿者——人生的意义不在于索取,而在于奉献!相信再小的爱心也能带来改变。
欢迎捐献造血干细胞——帮助千千万万的患者,改变别人的命运,只在点滴的善举。
欢迎成为新阳光的合作伙伴——合作共赢,让世界变得美好一点点。
欢迎给我们捐款——世界的改变不是少数人做了很多,而是每个人都做了一点点!

我们的生命,需要交流,需要人与人之间细微渺小但真实美好的关怀与爱。或促膝长谈,或细语言笑,或对酒当歌,或对床夜语。与志同道合的朋友为伴,它使我们的生命从荒芜走向繁盛,从凋零走向丰饶。您可以通过微博、微信公众号获得新阳光基金会的最新动态,与我们互动。同时可以在新阳光社区中,与更多专家、朋友进行交流与分享。
1.微信端,请点击右上角选择分享本文
2.浏览器中,请选择分享功能分享本文
【时 间】2018-01-31 20:00-21:00
【参与人次】截至发稿统计 472 人
【主讲嘉宾】张如艳,北京肿瘤医院乳腺肿瘤内科主治医师,博士,毕业于北京大学医学部(八年制),于核心期刊发表论文数篇。社会任职:中国女医师协会乳腺疾病中心秘书长。
【文字整理】李光启 黎虹宏 王丹迪
前言
姐妹们大家好,今天的微课堂将在微信群里开展,这个群是我们的主会场。今天的主题是关于临床试验的,这个主题我很早就想请专家来讲,但是没有机会,这次我们邀请到北京大学肿瘤医院乳腺内科主治医师张如艳大夫给大家分享关于临床试验的一些基本内容。
可能很多姐妹对临床试验有误解,也有很多姐妹私下问我临床试验怎么样、该怎么加入等等一系列的问题。我们知道临床试验其实就是在一个新研制的药品上市之前,经过动物、人体试验之后,才开展的Ⅰ期、Ⅱ期、Ⅲ期临床试验。临床试验的资质是很严格的,毕竟涉及到人身安全,不是随便哪个机构都可以开展临床试验,国家有资质的规定,也有法律、法规的限制,比如单中心、多中心联合。那这些都是什么意思呢?Ⅰ期、Ⅱ期、Ⅲ期之间到底有什么区别呢?这次我们就请张大夫专门给大家讲一下。大家知道临床医生都是特别忙的,张大夫也是用自己的休息时间给大家分享这个话题,机会非常难得。

香七微课堂的患者朋友们大家晚上好,我是北京肿瘤医院乳腺内科的张如艳医生,感谢香七微课堂提供这样一个机会和平台来进行医患之间的线上交流。今天我为大家分享的是临床试验或者说临床研究的内容。
一、药物临床试验知多少

我想很多患者对临床试验都有所了解,可能有一些患者已经参加了临床试验,但是还有一些患者对临床试验缺乏了解,或者存在一些错误的理解,今天主要从医学的角度来跟大家普及一些这方面的知识。
有些患者对临床试验不太了解,但是我想所有患者对药物说明书都很熟悉,说明书上的信息中,我们比较关注的可能有:适应症、药物用法用量、药物的不良反应,以及服药期间的注意事项等等。
其实,药品说明书上的这些信息——这里指的是西药——很多都不是凭空得来的,所有的信息都是通过严谨的临床研究得出来的。所以这也是临床试验存在的意义。临床试验主要的目的,就是评价新药潜在的临床应用价值,包括它的安全性、有效性等。通过临床试验来确定新药的使用方法,从而确定这个药的适应症,也就是最有可能从这个药物中获益的人群,同时通过临床试验来获得这个药物最佳疗效和最小不良反应的合适剂量。
二、药物临床试验的分类与分期
临床试验根据不同的特性、根据参加试验的中心数量、是否具有对照组、是否随机、是否有盲法等等,有不同的分类。根据临床试验的不同阶段,以及试验的主要目的,又可以分为Ⅰ期、Ⅱ期、Ⅲ期、Ⅳ期等不同的分期。有一些已经参加过临床试验的患者,在签署知情同意书的时候可能会发现标题大多是:多中心、随机、对照的Ⅲ期临床研究,其中也包含了一些对临床试验的分类和分期的信息。
临床试验从Ⅰ期到Ⅳ期的主要目的是不同的。Ⅰ期的试验是进行临床药理学的研究,主要是探索药物的安全性;Ⅱ期是初步评价药物的有效性;Ⅲ期是对药物的有效性进行确证;Ⅳ期的时候可能药物已经上市了,这个阶段主要是进行上市之后的一些监测,观察会否有一些罕见的不良反应。

Ⅰ期临床试验基本上是已经通过了动物试验,首次在人体中进行的安全性的试验,所以Ⅰ期临床试验的样本比较少,主要是健康的受试者或者患者。试验分为不同的剂量组,有时只是单次给药,测试患者药代动力学的一些指标,比如血药浓度,PK血等。也有多次给药的,比如每天给药,给到28天,然后在这期间不同的时间给病人采取不同的PK血。
所以一般Ⅰ期临床试验的患者是需要住进病房来的,因为需要抽血的次数比较多,比较频繁,而且需要密切监测不良反应。有的中心有专门的Ⅰ期临床病房,有的就是普通病房,但是实际上对患者的观察是一样的。
在Ⅰ期临床试验中得出比较好的安全数据之后,就可以在此基础上扩大样本量进行Ⅱ期的药物临床试验,在继续评价药物安全性的基础上,初步评价药物对患者的治疗效果,从而为Ⅲ期临床试验的研究设计和给药剂量方案提供依据。

在Ⅱ期临床试验结束后,基本上已经有药物的安全性,初步治疗效果,以及一个比较合适的给药剂量和给药方案,此时就可以进行扩大的、多中心、随机、对照的、拥有足够样本量的Ⅲ期临床试验。
Ⅲ期临床试验是药物上市前最重要的一个步骤,主要是进一步评价药物对目标适应症患者的有效性和安全性,评价利益与风险关系,为药物注册上市申请获得批准提供充分的依据。
其实有些药物在国外已经上市了,但是在国内的患者中还没有进行相关的临床试验,所以一般会首先在中国的患者当中进行Ⅱ期的临床试验,这个时候往往是没有随机分组的,可能直接就进入到试验组中。
在试验组中得到了很好的安全性以后,就可以进入我们称之为“在中国的注册研究”阶段,即在中国的Ⅲ期临床试验。比如我们科正在开展的,Ⅲ期国际多中心的,关于HER-2阳性、赫赛汀治疗失败的应用TDM1治疗的一个药物临床试验。
Ⅳ期药物临床试验主要是针对一些已经上市的药物,监测它比较罕见的一些不良反应或者是在特殊人群当中的不良反应,所以它是上市后的药物监测。一般是由研究者或者医生自主发起的临床研究阶段。
临床试验不管是Ⅰ期还是Ⅳ期,从研究的申请到开展其实是需要经历一个非常复杂的步骤的,首先需要申办方向中国药监局提交临床试验的申请,在获得药监局对临床研究的批准之后,拿到临床研究的批件,才能开始准备相关的资料。完成之后,他们会选择信任的研究中心或者研究者来进行研究,并由研究者来制定一些入选标准和排除标准。
最终批下来的这些研究方案和执行同意书等内容,还要交给研究中心的伦理委员会进行审批。伦理委员会主要是为了保护受试者的利益成立的一个委员会,里面包括医学专业的人员,律师等法律方面的人员,还有与医学不相关的代表患者的人群。所以有时候你可能听说某个试验要开展了,但是其实从听说到真正启动,可能会经过几个月、半年乃至一年的时间。
临床试验启动之后,才真正开始筛选患者,进行患者的招募。临床研究启动之后,我们会在门诊或者病房挑选条件适合的患者,跟她们谈谈试验相关的信息。比如:我觉得你正常情况下应该接受的治疗方案是怎样的,目前有什么样的临床试验是适合你的……我们会向患者说明相关情况,如果患者有这方面的意向,就可以签署知情同意书。签了知情同意书之后,才算真正的参加了临床试验,正式进入了这个包括筛选期、治疗期和随访期的临床试验。
在这里需要跟患者们强调一下,临床试验这个过程是通过你自己考虑的、完全自愿的过程,如果有任何疑问,都可以跟你的研究者提出。如果对哪方面产生了疑虑,不想参加这个试验,是完全可以的,而且也不会影响你的正常治疗,如果不参加这个试验的话,我们肯定还会有临床常规的治疗方案提供给患者。

签署知情同意书之后不是直接入组到临床试验,而是要进入筛选期,完善一些相关的检查。临床试验有一些入组标准和排除标准,在完善检查之后,满足所有的入组标准,并且不符合所有排除标准,才能称为筛选成功,进入到临床试验里来。
筛选期也是为了保障获益人群,而且患者需要在入组前具有良好的器官功能,能耐受试验的药物,排除禁忌症。一个是评价患者的器官功能,比如血常规,白细胞、血小板,是否有贫血,心脏功能是否良好,心电图是否正常,是否有活动性的隐形肝炎等这些有可能影响试验药物的情况,这是患者身体的基本情况。还有患者肿瘤负荷的情况,因为Ⅲ期的药物试验评估药物的疗效,所以需要指导患者治疗前的状态,和治疗后的肿瘤状态进行对比。
有些患者在试验前曾服用过其他抗肿瘤的药物,为了排除其他药物的影响,一般需要经过一个停药期——我们称为洗脱期——来把这个药物的效果排除。一般需要21天-28天的时间,有些研究中对内分泌治疗的洗脱时间的要求可能会缩短到一到两周,主要是根据药物的半衰期来决定的。
洗脱期往往是和筛选期重合的,一边停药,一边进行筛选,以免因为停药时间过长造成患者疾病进展的情况。
在所有的检查完成之后,患者符合所有的入组标准,不符合所有的排除标准,就算筛选成功,这时就可以进入治疗期。大部分试验是随机分组的,有可能被分到试验组,也有可能被分到对照组,对照组一般是临床常规的治疗药物,一般是按照1:1、2:1、3:1等的不同比例来进行。治疗期间根据试验方案来用药,定期进行毒性检测和肿瘤疗效的评估,毒性检测主要包括我们刚才提到的一些血项、肝肾功能、心脏毒性等等。
如果在治疗结束以后,这个药物对患者依然有效的情况下,将会进入到随访期。在随访期患者依然会接受定期的复查,然后到门诊、病房进行访视,主要是观察用药以后的安全情况,以及疗效的情况。随访的方式有时通过电话随访,也有时候可能患者出现了不适的症状,需要到门诊去就诊,这也是随访的其中一种形式。
三、临床试验中不同角色、分工
在临床实验的流程当中,是需要很多人来参与的,除了主要研究者之外,下边一般会有负责临床研究的医生,称为助理研究者,对患者进行筛选,主要负责患者的入排、签署知情同意书。还会有临床研究协调员,我们叫CRC,来协助患者进行检查的预约、制订随访时间等等细致的事情。还有负责抽取PK血、给药的研究护士。还有患者进行影像检查之后需要有一个固定的医师来进行肿瘤评估,以保证整个临床研究的过程的严谨性。
四、其他
一个设计良好的临床试验主要是从患者出发,是为受试者的利益考虑而设计出来的一个临床试验。尤其很多肿瘤患者,在常规抗肿瘤情况下,疗效不是很理想的情况下,通过临床试验也许能获得意想不到的效果。
在乳腺癌领域非常权威的一个指南,叫《美国国立综合癌症网络NCCN乳腺癌临床试验指南》,在它的首页中明确指出,任何肿瘤患者都可以从临床试验中得到最佳的处理,因此它特别鼓励肿瘤患者参加临床试验。


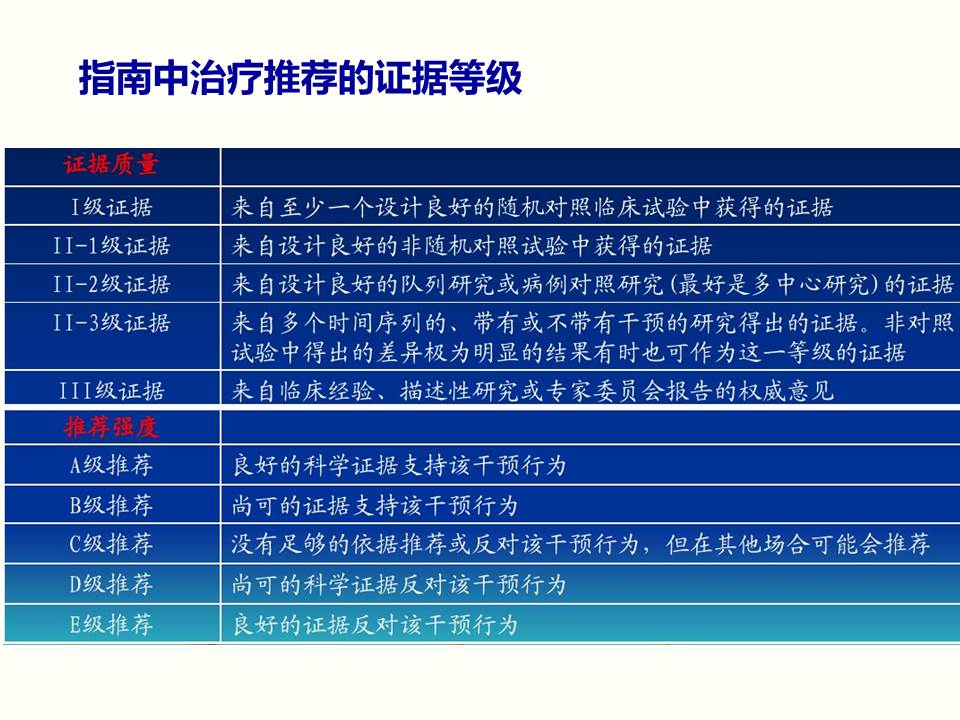
在《指南》当中,每给出一条治疗的推荐,后边括号里都会标注出推荐的证据等级是什么。证据等级由上到下,最高级的1-A级的证据主要是来自一个设计良好的随机对照的临床试验中,证明这是有良好的科学证据支持该干预行为。
最后想多说两句,不管是临床当中推荐患者入组到临床试验里来,还是给患者进行的一些抗肿瘤的治疗决定,其实这背后都是我们定期对患者的病情进行详细分析之后——有时还是各个专家一起商量出来——的决策,主要目的都是希望患者能够获得一个比较良好的抗肿瘤的效果。
医患双方实际上是在同一个战壕里的战友,我们具有共同的敌人、共同的目标。所以希望我们医患双方能够共同努力,攻克肿瘤,也希望各位患者都能取得比较好的治疗效果。
温馨提示
微课堂提供的资源,仅用于教育和信息目的,所提供的信息为一般性内容。如想针对特定治疗问题或关心的内容获取答案,请详细咨询主治医生或专业人士。该内容不得以任何方式替代专业咨询或医疗咨询。



扫描二维码
关注新阳光微信公众号


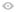












 返回新阳光首页,查看更多
返回新阳光首页,查看更多




